Jeanne M. Frederick

Research Associate Professor of Ophthalmology & Visual Sciences
Molecular Neuroscience
Neurobiology of Disease
e-mail: jeanne.frederick@hsc.utah.edu
Ph.D. 1979, University of Wisconsin-Madison School of Medicine Madison, WI; Postdoctoral
Fellow, 1979-1982, Baylor College of Medicine Houston, TX
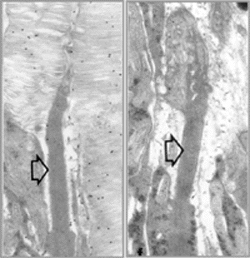
Ultrastructure of P15 mouse retinas, comparing normal rod photoreceptor (left) to
a rod expressing a mutant rhodopsin (right). Open arrows indicate connecting cilia
for reference. Gold particle labeling of normal retina reveals rhodopsin localization
in rod outer segment membrane. Retinas expressing a triple mutant rhodopsin transgene
in the absence of normal rhodopsin do not form rod outer segment membranes, suggesting
that the mutant protein cannot substitute for its normal counterpart. Rather, the
mutant protein folds incorrectly and is retained in the endoplasmic reticulum.
RESEARCH:
GOAL: To investigate the pathogenesis of mutant protein expression in retina neurons
We study the organization of the retina in health and disease. Toward this end, the phenotypes of transgenic and knockout/ knock-in mice carrying mutations in genes linked to retina dystrophies are analyzed. We are particularly interested in signal transduction, remodeling of the inner retina subsequent to rod and cone photoreceptor degeneration, and mechanisms that lead to cell death. In several neurodegenerative diseases, unfolded proteins accumulate intracellularly as insoluble inclusions and may serve a critical role in disease progression. If native polypeptide conformations are lost through genetic mutation or postsynthetic damage (e.g., environmental stress), cells normally have elaborate mechanisms to prevent the aggregation of unfolded proteins, to attempt refolding and, if proper folding is impossible, to degrade the abnormal polypeptides into amino acids.
However, we have shown that in a mouse model for human adRP (autosomal dominant retinitis pigmentosa), expression of a mutant rhodopsin (visual pigment) generates a protein that fails to mature, transport and support normal rod photoreceptor outer segment formation and, moreover, is cytotoxic. The mutant protein folds incorrectly and is retained in the endoplasmic reticulum thereby initiating an unfolded protein response. What is the link between expression of a misfolded, mutant protein and induction of cell death? To address such questions, we employ gene cloning, bright-field and confocal microscopy and related techniques in cell and molecular biology. For an example, see the recent publication: